
Il 30 agosto u.s. Bayer ha annunciato[1] che la Food and Drug Administration (FDA) statunitense ha approvato il BAY94-9027 (Jivi®), fattore antiemofilico ricombinante peghilato ( damoctacog alfa pegol), per il trattamento dell’emofilia A negli adulti e negli adolescenti, precedentemente trattati, di età pari o superiore ai 12 anni [1,2] .
Tweet Bayer US del 31 Agosto 2018
Il regime profilattico suggerito, grazie all’emivita prolungata di Jivi®, consente di raccomandare inizialmente una somministrazione bisettimanale che può essere rimodulata ogni cinque giorni e ulteriormente adattata per ogni soggetto con frequenza variabile più o meno prolungata in base agli episodi di sanguinamento. La FDA ha inoltre approvato Jivi® per il trattamento on demande per la prevenzione perioperatoria del sanguinamento nella stessa popolazione di pazienti.
La richiesta per l’autorizzazione all’introduzione nella pratica clinica del BAY94-9027, damoctacog alfa pegol, era stata presentata nel novembre 2017 sulla base dei risultati positivi del trial di fase 2/3 PROTECT VIII .
In questo studio multinazionale di fase 2/3, aperto e parzialmente randomizzato sono stati arruolati emofilici A con FVIII <1% di età compresa tra 12 e 65 anni con almeno 150 giorni di esposizione al Fatt.VIII. La sicurezza e l’efficacia del damoctacog alfa pegol , fattore VIII ricombinante a lunga emivita, sono stati valutati dopo 36 settimane di esposizione allo stesso.
BAY 94-9027 è una molecola di FVIII ricombinante B deleta peghilato con una singola molecola di PEG da 60 kDa associata a un residuo di cisteina ingegnerizzato [5,7,8].
In uno studio di fase I (NCT01184820), che arruolava 14 soggetti con emofilia A grave, il BAY 94-9027 aveva mostrato un’emivita di circa 19 ore; il 50% in più rispetto al r-FVIII formulato con saccarosio con un recupero comparabile [3].
E’ sulla base dei risultati di questa prima fase che è stato quindi progettato ed eseguito il PROTECT VIII con l’obbietivo di valutare appunto l’efficacia e la sicurezza di BAY 94-9027 in 134 pazienti con emofilia A grave precedentemente trattati con altri prodotti [4]. Come outcome primario di efficacia quindi il Trial individuava il tasso di sanguinamento annualizzato (ABR).
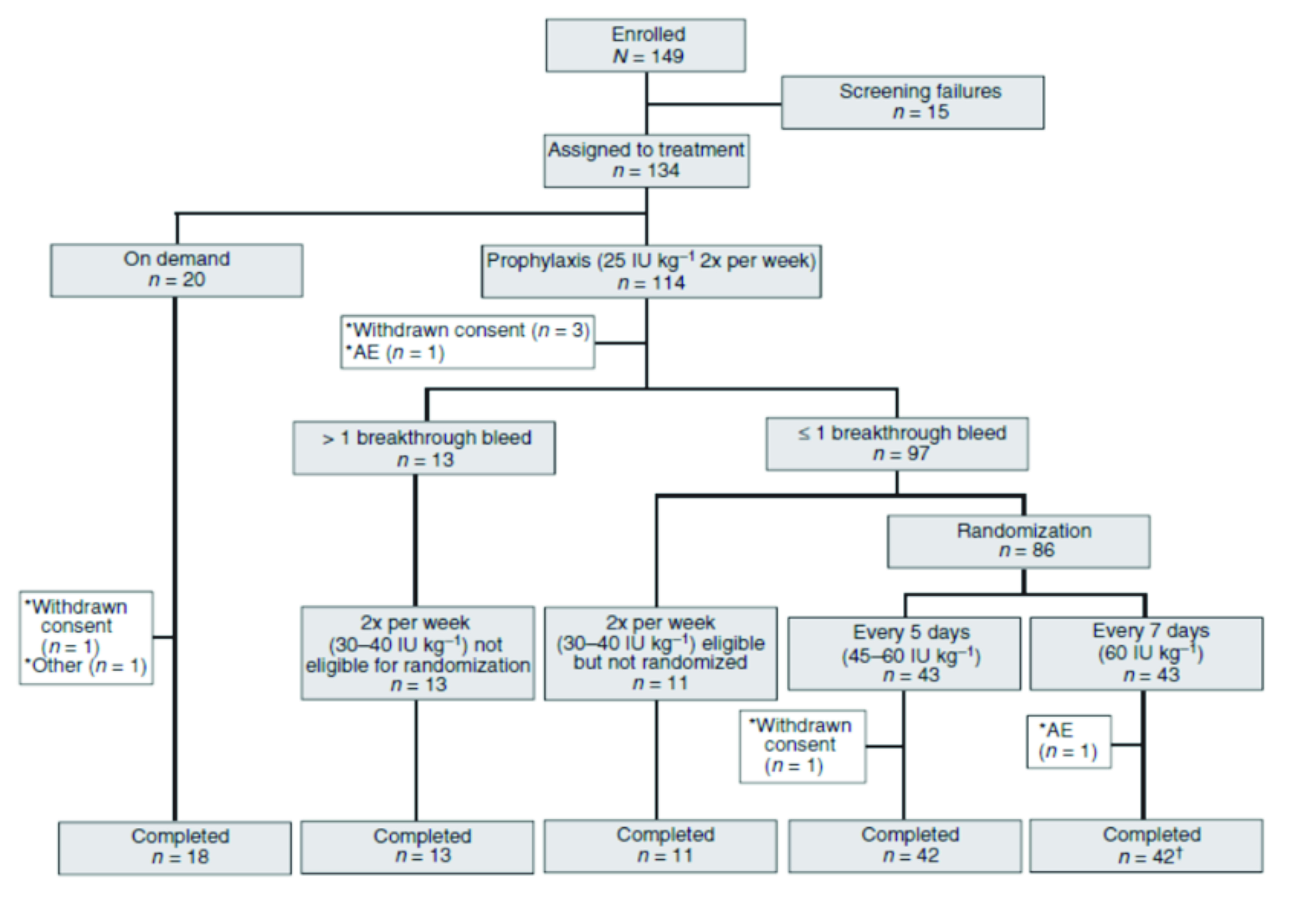
Lo schema di arruolamento tratto da M.T.Reding et al., Journal of Thrombosis and Haemostasis, 15: 411–419,2016
Lo studio ha previsto che i pazienti in profilassi, prima dell’ingresso nello studio, erano elegibili solo per essere arruolati con un regime terapeutico di profilassi, mentre quelli precedentemente trattati on demandpotevano continuare con un regime a domanda o in alternativa scegliere di entrare nel braccio di profilassi.
I pazienti entrati nel braccio di profilassi (n.114) sono stati inizialmente trattati con 25 u.i./kg bisettimanali di BAY 94-9027 durante un periodo di osservazione preliminare di 10 settimane. Periodo che è stato utilizzato per identificare pazienti che avevano una tendenza al sanguinamento più frequente e per i quali era prevedibile che non potessero utilizzare un regime terapeutico con infusioni più dilazionate.
Questi ultimi (n.18), definiti a rischio, hanno ricevuto un aumentato della dose a 30/40 u.i./Kg, non sono stati elegibili per una randomizzazione e hanno continuato con un regime terapeutico di due infusioni settimanali.
I pazienti, con un buon controllo del sanguinamento (≤ 1 episodio) nel periodo di osservazione, sono stati poi randomizzati 1: 1 per ricevere BAY 94-9027 ogni 5 giorni (a partire da 45 IU kg) o ogni 7 giorni (dose fissa di 60 UI kg) per 26 settimane. I due bracci di randomizzazione non hanno raccolto tutti i pazienti elegibili ma solo un numero sufficiente a completare la composizione degli stessi limitata a 43 pazienti ciascuno. I rimanenti 11 pazienti, elegibili ma non randomizzati, hanno continuato con un regime terapeutico di profilassi bisettimanale.
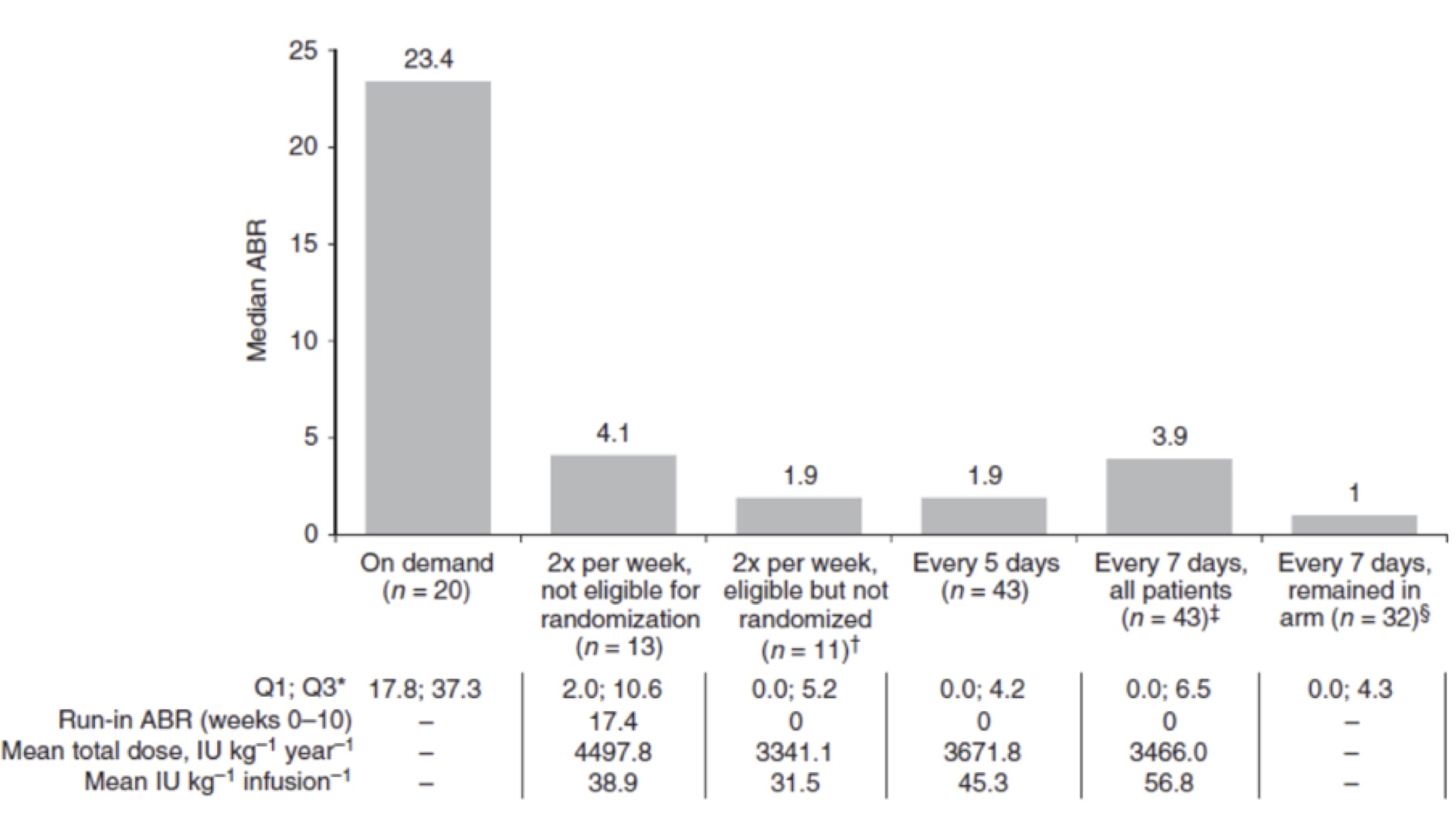
Episodi emorragici e dosaggi, ABR, indice annualizzato di sanguinamento (annualized bleeding rate); Q1, quartile 1; Q3, quartile 3.
tratto da M.T.Reding et al., Journal of Thrombosis and Haemostasis, 15: 411–419,2016
Nelle prime 10 settimane di osservazione tutti i pazienti elegibili per la randomizzazione hanno mostrato una mediana di ABR pari a 0.0 sia per quelli inseriti nel braccio con trattamento a 5 giorni che per quelli in 7 giorni. I pazienti elegibili ma non inseriti nei due bracci di cui sopra hanno anch’essi mostrato lo stesso ABR 0.0. Al contrario i pazienti non elegibili, sempre nello stesso periodo, e in trattamento bisettimanale hanno mostrato un ABR 4.1.
Nelle settimane successive sino alla 36°, in seguito all’incremento della dose mediana (39.0 u.i./kg) i pazienti non elegibili hanno comunque migliorato il loro ABR.
I pazienti randomizzati nel gruppo con trattamento ogni 5 giorni hanno mostrato un basso ABR di 1,9. Comportamento identico si è osservato anche nei pazienti elegibili ma non randomizzati. In tutti e due questi gruppi si è potuto registrare rispettivamente il 46% ed il 44% di pazienti senza alcun episodio emorragico.
Nel braccio che accoglieva i pazienti trattati ogni 7 giorni l’ABR ha segnato 3.9. L’ABR è stato calcolato per tutti i pazienti arruolati in questo gruppo e per tutto il tempo che essi sono rimasti in questo braccio. Il 25% dei pazienti trattati ogni sette giorni ha presentato ≥1 sanguinamento e la loro frequenza di somministrazione è stata portata a cinque giorni; d’altra parte, nessuno di questi pazienti ha poi avuto necessità di aumentare la frequenza di somministrazione [04].
Il 75% dei pazienti, invece, che è rimasto stabilmente nel braccio ha realizzato un ABR di 0.96 e il 50% di essi non ha presentato alcun sanguinamento.
In sintesi l’ABR mediano nella profilassi era 4,1 nei pazienti non elegibili trattati due volte a settimana, 1,9 in tutti quelli, elegibili e non, trattati ogni cinque giorni e 0,96 in quelli che potevano mantenere un intervallo di trattamento di sette giorni [04]. Il 91% degli episodi di sanguinamento sono stati controllati con una o due infusioni.
Lo studio ha sicuramente raggiunto il suo obbiettivo primario che era la prevenzione del sanguinamento mediante terapia somministra ad intervalli sino a 7 giorni. Una effettiva e significativa profilassi con BAY 94-9027 è stata sicuramente dimostrata con la somministrazione bisettimanale e con quella ad ogni 5 giorni. Per altro nel 75% dei pazienti arruolati nel braccio ad intervalli di 7 giorni la somministrazione settimanale è stata pienamente efficace nel controllo dell’emostasi. Anche in questa esperienza si rafforza l’osservazione che, conoscendo la storia personale dei pazienti, è possibile identificare il regime terapeutico adatto per ogni individuo e nella fattispecie l’intervallo di somministrazione più adatto.
Ultima nota da sottolineare è che nessun paziente ha sviluppato anticorpi neutralizzanti anti-FVIII [04]. Attualmente è in corso uno studio multicentrico di fase 3, in aperto, non randomizzato per valutare l’efficacia e la sicurezza di BAY 94-9027 nei bambini precedentemente trattati con emofilia grave A di età <12 anni (NCT01775618, PROTECT VIII Kids) [6]. Lo studio è stato recentemente completato ma i risultati finali non sono ancora disponibili.
RIFERIMENTI
01. https://www.bayer.us/en/newsroom/press-releases/article/?id=123236
02. Jivi® [prescribing information]. Whippany, NJ: Bayer; 2018.
03. Coyle T.E., Reding M.T., Lin J.C., Michaels L.A., Shah A., Powell J. Phase I study of BAY 94-9027, a PEGylated B-domain-deleted recombinant factor VIII with an extended half-life, in subjects with hemophilia A. J. Thromb. Haemost. 2014;12:488–496. doi: 10.1111/jth.12506.
04. Reding M.T., Ng H.J., Poulsen L.H., Eyster M.E., Pabinger I., Shin H.J., Walsch R., Lederman M., Wang M., Hardtke M., et al. Safety and efficacy of BAY 94-9027, a prolonged-half-life factor VIII. J. Thromb. Haemost. 2017;15:411–419. doi: 10.1111/jth.13597.
05. Mei B., Pan C., Jiang H., Tjandra H., Strauss J., Chen Y., Liu T., Zhang X., Severs J., Newgren J., et al. Rational design of a fully active, long-active PEGylated factor VIII for hemophilia A treatment. Blood. 2010;116:270–279. doi: 10.1182/blood-2009-11-254755.
06. Santagostino E., Saxena K., Kenet G., Fischer K., Biss T., Radke S., Michaels L.A. PROTECT VIII Kids trial results: BAY 94-9027 safety and efficacy in previously treated children with severe hemophilia A. Haemophilia. 2016;22:41. (www.postersessiononline.eu/173580348_eu/congresos/WFH2016/aula/-MPT_72_WFH2016.pdf)
07. A. Shah T. CoyleS. LalezariK. FischerB. KohlstaeddeH. DelesenS. Radke L. A.Michaels: BAY 94-9027, a PEGylated recombinant factor VIII, exhibits a prolonged half-life and higher area under the curve in patients with severe haemophilia A: Comprehensive pharmacokinetic assessment from clinical studies . Haemophilia. 2 018;1–8 . DOI: 10.1111/haem.13561
08. Mei B, Pan C, Jiang H, Tjandra H, Strauss J, Chen Y, Liu T, Zhang X, Severs J, Newgren J, Chen J, Gu JM, Subramanyam B, Fournel MA, Pierce GF, Murphy JE. Rational design of a fully active, long-acting PEGylated factor VIII for hemophilia A treatment. Blood 2010; 116: 270–9.